Steering a battery manufacturer’s application through the U.S. FDA validation process
There is an extensive and rigorous process by which a battery for a medical device achieves validation in compliance with FDA regulations
BY JULIA PALU
VARTA Microbattery
Power Pack Solutions
www.varta-microbattery.com
A recent change in outlook at the U.S. Food and Drug Administration (FDA) — the government body that regulates the use of devices and substances in medical care — has escalated the already rigorous tests applied to medical devices to new, higher levels.
In 2009, FDA Commissioner Margaret Hamburg told medical device manufacturers that they bear responsibility for every step in their supply chain on questions of product under-performance or malfunction. “Whether your supplier is on the other side of the world, in this hemisphere or right next door, you are responsible,” she said. In 2009, 12% of the observations and 16% of the warning letters issued by the FDA were related to inadequate supplier qualifications.
The message is clear for medical device manufacturers, and for the suppliers who provide them with components. It is not enough for end product manufacturers to prove the efficacy of their product — they must also independently validate the performance of key components.
A battery is normally regarded as a key component, for two reasons:
As the power source for portable devices, the battery is mission-critical. If the battery suffers unforeseen catastrophic failure or premature depletion, every other component in the system will be disabled.Batteries can be completely safe, but only if control, charging and protection techniques are correctly applied.
This article describes the extensive and rigorous process by which a battery for a medical device achieves validation in compliance with FDA regulations. There are important aspects to be considered by makers of battery-powered devices:
1. Instituting the processes required in the FDA’s regime takes a long time and a vast amount of engineering and management effort. For a battery manufacturer embarking on FDA validation for the first time, this could have an impact on the product design process and on product introduction timescales.
2. The rigour and depth of the validation process brings to light new potential for design and production improvements. Customers across the board are benefiting from the improvements that VARTA Microbattery has implemented as a result of undergoing the FDA validation process.
The FDA validation regime
The FDA is emphasizing the importance of complying with supplier controls in an effort to stem the rising number of recalls of commercial products. The controls are backed by an extensive program of audits. This audit regime has teeth: the FDA frequently issues “citings,” which require immediate remedial action on the supplier’s part.
The FDA process is concerned with checking the attributes of a product or component that, if not controlled, can affect safety, quality, performance or efficacy. Control is effected through verification (performance testing of finished product — not samples, but every single unit); or, if verification is not possible, through validation. Validation procedures are designed in accordance with the FDA’s standard CFR820, and with ISO 9001 and ISO 13485.
It would be unreasonable to expect device manufacturers to validate every single attribute of every single component. In practice, manufacturers will make a risk assessment, and prioritize processes and components in order of risk.
In the case of batteries, many important attributes cannot be verified; examples would include charge capacity decline over time, and resistance to shock. In both cases, verification testing would invalidate or damage the unit being verified, rendering it unfit for sale.
So most important attributes of a battery need to be validated rather than verified. The purpose of validation is to show, with appropriate documentation, that:
• Manufacturing processes are designed so as to produce a component that performs in accordance with its specification
• These processes are implemented consistently and correctly
• In order to achieve validation for a battery in a medical device, a battery manufacturer must be able to show:
An evidence-based rationale for the Design of Experiments technique used to prove the effectiveness of processes employed in the production of a batteryAn evidence-based rationale for the parameters used to assess performanceWithin these parameters, an evidence-based rationale for the setting of the values that define the acceptable and unacceptable performance
This can be illustrated by way of an example. Let us assume that one important attribute of a portable medical device should be tolerance of shock. Since shock presents a serious risk to the proper functioning of the battery, the attribute of tolerance of shock must be validated in the battery as well as the end product it is built into.
The battery manufacturer uses the DOE technique to identify the optimum operating range for all design parameters leading to successful experiments and evidence that the battery’s tolerance of shock satisfies the requirements. A drop test — for example, dropping the battery to the ground from a height of 1 m — could be used to test for tolerance of shock. The battery manufacturer must also set pass/fail values for the test: the battery must survive n number of drops without damage to the casing and without any degradation of performance.
The FDA standards require technical documentation that proves the rationale for selecting the test method, the critical parameters, and the optimum range, and this documentation must be capable of withstanding scrutiny. Is it the best possible test of tolerance of shock? Could a different test produce a more reliable measure of the battery’s tolerance of shock? If not, why not?
The battery manufacturer must also be able to justify the pass/fail value for the parameter that is being measured. In this case, the documentation must show why n drops is the right value for a Pass in the drop test. Why would n + 10 or n – 5 not be better thresholds for acceptable performance in relation to tolerance of shock? How can the battery manufacturer be sure that a battery that survives n drops in the test meets the requirements of the product specification?
Let us assume that surviving n drops in the 1-m drop test is accepted as a proper measure of tolerance of shock. Now the battery manufacturer has to validate the assembly process that will produce in volume batteries that will, if tested, pass the drop test. Again, validation involves a series of steps (see Fig. 1 ), at each of which the battery manufacturer must devise and justify a protocol, and then show that it executes the protocol consistently.
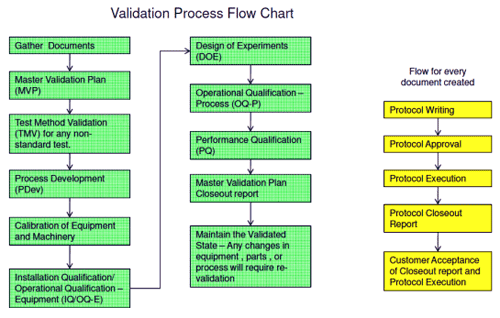
Fig. 1: Typical FDA validation process flow applied to a battery pack for use in a medical device
Installation Qualification (IQ) (see Fig. 2 ) checks factors such as installation conditions, documentation, the equipment’s design and the environmental conditions in which it operates. IQ leads on to OQ-E (Operational Qualification of Equipment), which checks for correct operation of the equipment. This validates the equipment’s functionality, operating procedures, materials handling procedures, and even the training of the machine operators.
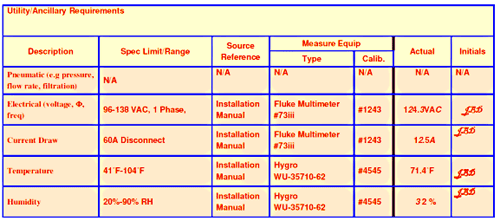
Fig. 2: Typical section of IQ requirements document
When the installation and operation of the production equipment have been qualified, the actual manufacturing process itself is validated in operational qualification — process (OQ-P). In the example of tolerance of shock, this might for instance require validation of the Ultrasonic Welding method for the plastic housing, in order to optimize parameters such as the amplitude, force applied and distance to target of the welding equipment.
Performance qualification (PQ) then checks that the correct implementation of the process validated under OQ-P does in practice produce batteries that meet the specification. PQ looks for potential variations attributable to, for instance, staff/shift changes, and variations in components.
Validation effect on battery manufacturers and customers
VARTA Microbattery has already piloted a battery through the FDA validation process; it is, without doubt, exhaustive, detailed, time-consuming and expensive the first time it is attempted. The lessons learned from it and the investments made will allow VARTA to accelerate the process in the future. For their part, medical device manufacturers need to be aware of the complexity of the battery validation process, to allow for it in their product marketing plans, and to set aside time to collaborate with suppliers on the design and execution of validation protocols.
Interestingly, the FDA validation process also exposed VARTA Microbattery to a more intense, detailed focus on the testing and optimisation of every step in the battery production process than is found anywhere else in the electronics industry. VARTA, which benchmarks itself against the world’s other top battery manufacturers and aims for industry-best quality standards, found through the FDA validation process that it was able to improve both product design (for instance, in relation to materials used) and product assembly processes (for instance, in relation to ultrasonic welding techniques) as a result of implementing the procedures required by the FDA regime. ■
Advertisement
Learn more about Varta Batteries




